For healthcare professionals
Myositis
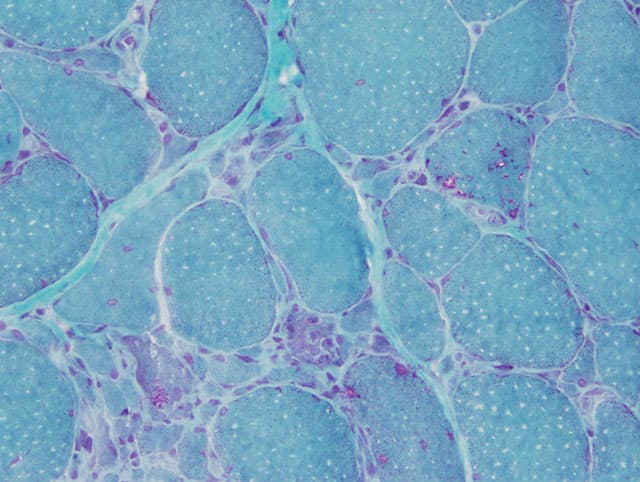
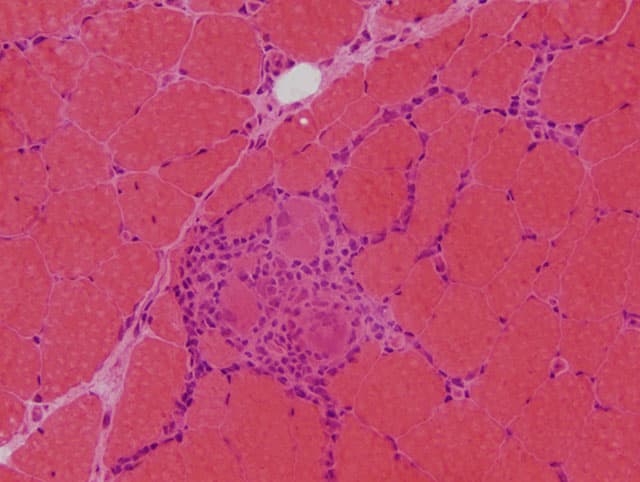
Myositis – also called inflammatory myopathies– comprises a group of diseases that are often challenging for patients as well as for physicians. They are characterized by proximal weakness of arms and legs and different degrees of extramuscular affection including symptoms of the skin, lung or joints. Several forms of myositis can be associated with cancer. The diagnosis requires a combined assessment of the clinical symptoms, presence of auto-antibodies and pattern of muscle histopathology. The current classification distinguishes the following subtypes of myositis:
Overlap Myositis (OM)
This group is characterized by the presence of distinct antibodies such as antisynthetase antibodies (most frequently anti-Jo1, anti-EJ, anti-OJ and others), anti-Ro52, and anti-Ku. An overlap myositis can occur in presence of other distinct autoimmune diseases, such as systemic sclerosis, which can be associated with anti-PmScl-antibodies.
Antisynthetase Syndrome (ASyS)
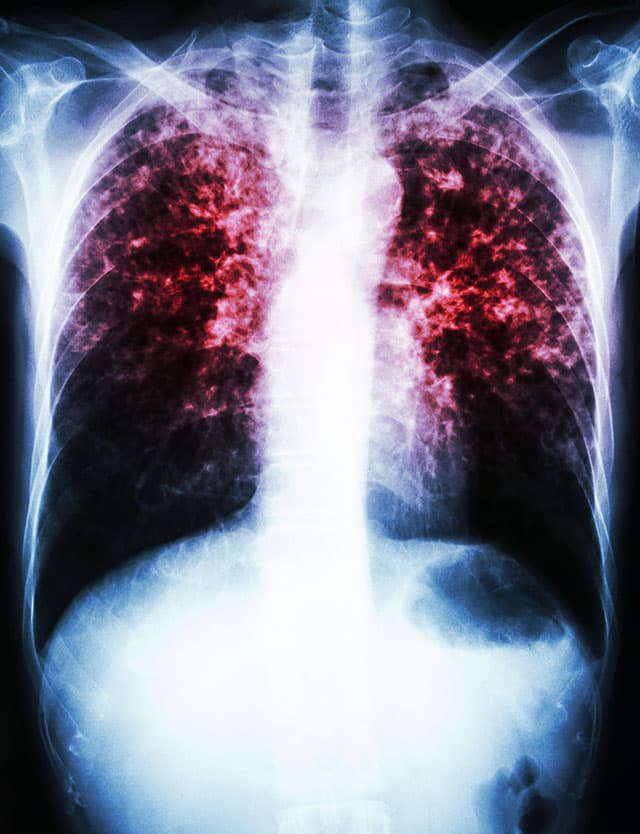
This is currently considered a distinct entity and characterized by the presence of fever, Raynaud’s phenomenon, arthritis, mechanic’s hands, interstitial lung disease, and myositis. Typically, not all organ manifestations are present concomitantly, but may develop over time. Antisynthetase antibodies such as Anti-Jo1, -PL7, -PL12, -KS, -OJ, -EJ, -Zo, -Ha; may be present.
Dermatomyositis (DM)
This form of myositis is characterized by the presence of symptoms of the skin including e.g. gottron’s papules or V-sign. In juvenile forms, subcutaneous calcification may occur. Classical dermatomyositis is often associated with anti-Mi2 antibodies. Particularly older patients or those with anti-TIF1γ antibodies are often associated with cancer. A distinct course of the disease is called “clinically amyopathic dermatomyositis (CADM)”, which often is associated with anti-MDA5 antibodies and typically includes a severe interstitial lung diseases.
Immune-mediated Necrotizing Myopathy (IMNM)
This type of myositis is characterized by severe weakness of arms and legs and often highly elevated muscle enzymes (creatine kinase or CK). It can be associated with the auto-antibodies anti-HMG-CoA receptor or anti-SRP. Some patients develop an interstitial lung disease and an association with cancer can be present.
Polymyositis (PM)
Polymyositis is characterized by proximal weakness of arms and legs. An association with unique auto-antibodies has not been identified. In view of the aforementioned myositis subtypes that can be associated with distinct auto-antibodies and characteristic patterns of muscle histology, polymyositis can be considered as the least frequent subtype of myositis.
Inclusion Body Myositis (IBM)
Inclusion body myositis is characterized by proximal and distal weakness of arms and legs and only mildly elevated CK. With an onset beyond 45 years of age, the disease is slowly progressive and often leads to an impaired ambulation. Many patients develop swallowing difficulties, which may cause malnutrition or aspiration. An anti-cN1A auto-antibody is observed in about a third of the patients.